
Therapiegebiete & Indikationen
Lungenfibrose
Leitlinien zur Diagnostik der IPF und anderer interstitieller Lungenerkrankungen
In den vergangenen Jahren sind zahlreiche internationale und nationale Leitlinien zur Diagnose und Therapie interstitieller Lungenerkrankungen (ILDs) veröffentlicht worden. Sie bieten strukturierte und evidenzbasierte Empfehlungen, die eine frühzeitigere und präzisere Diagnose ermöglichen. Sie tragen zudem dazu bei, Therapieentscheidungen zu optimieren und die interdisziplinäre Zusammenarbeit zu stärken. Dieser Beitrag bietet einen Überblick über
- die aktuelle S1-Leitlinie Interdisziplinäre Diagnostik interstitieller Lungenerkrankungen im Erwachsenenalter aus dem Jahr 20231 inkl. Foliensatz zum Download
- die Kriterien für den Nachweis einer progredienten Lungenfibrose2
- die Empfehlungen für das ILD-Screening und Monitoring der ERS/EULAR Leitlinie für die klinische Praxis bei interstitiellen Lungenerkrankungen im Kontext von Bindegewebserkrankungen (CTD-ILD) und rheumatoider Arthritis (RA-ILD).3
ILD-Diagnose inkl. IPF Schritt für Schritt – nationale S1-Diagnostikleitlinie1
Die frühzeitige korrekte Diagnose einer ILD-Entität ist für die Prognose und Therapie der Patient:innen entscheidend. Die S1-Leitlinie definiert die diagnostischen Schritte und trägt mit den strukturierten Handlungsempfehlungen praxisnah zu einer frühzeitigen Identifikation von ILD-Pati-ent:innen bei.
Bei klinischem Verdacht auf eine interstitielle Lungenerkrankung (ILD) sind folgende Kriterien zu berücksichtigen:
1. Anamnese und körperliche Untersuchung1
Eine sorgfältige Anamnese und körperliche Untersuchung mit Schwerpunkt auf Expositionen, Systemerkrankungen, Medikamenteneinnahme und Familienanamnese ist entscheidend.
Die S1-Diagnostikleitlinie empfiehlt die Verwendung eines standardisierten Fragebogens, um alle wesentlichen Aspekte zu berücksichtigen.

Typische körperliche Anzeichen/Symptome eine Lungenfibrose sind: Sklerosiphonie bei Fibrose/inspiratorisches Juchzen bei exogen allergischer Alveolitis (EAA) bei Auskultation der Lunge, Trommelschlegelfinger, Charakteristika einer Systemerkrankung/ Sarkoidose, Rechtsherzdekompensation
2. Lungenfunktionsdiagnostik1
Die Durchführung der folgenden lungenfunktionellen Prüfungen wird empfohlen:
- Spirometrie/Bodyplethysmografie
- Diffusionskapazitätsbestimmung
- Blutgasanalyse (BGA)
- Belastungstests (6-Minuten-Gehtest/Spiroergometrie)
3. Native HRCT1
Die Durchführung einer nicht kontrastmittelverstärkten dünnschichtigen Spiral-Computertomografie des Thorax ist unerlässlich.
Folgende HRCT-Einstellungen werden empfohlen1

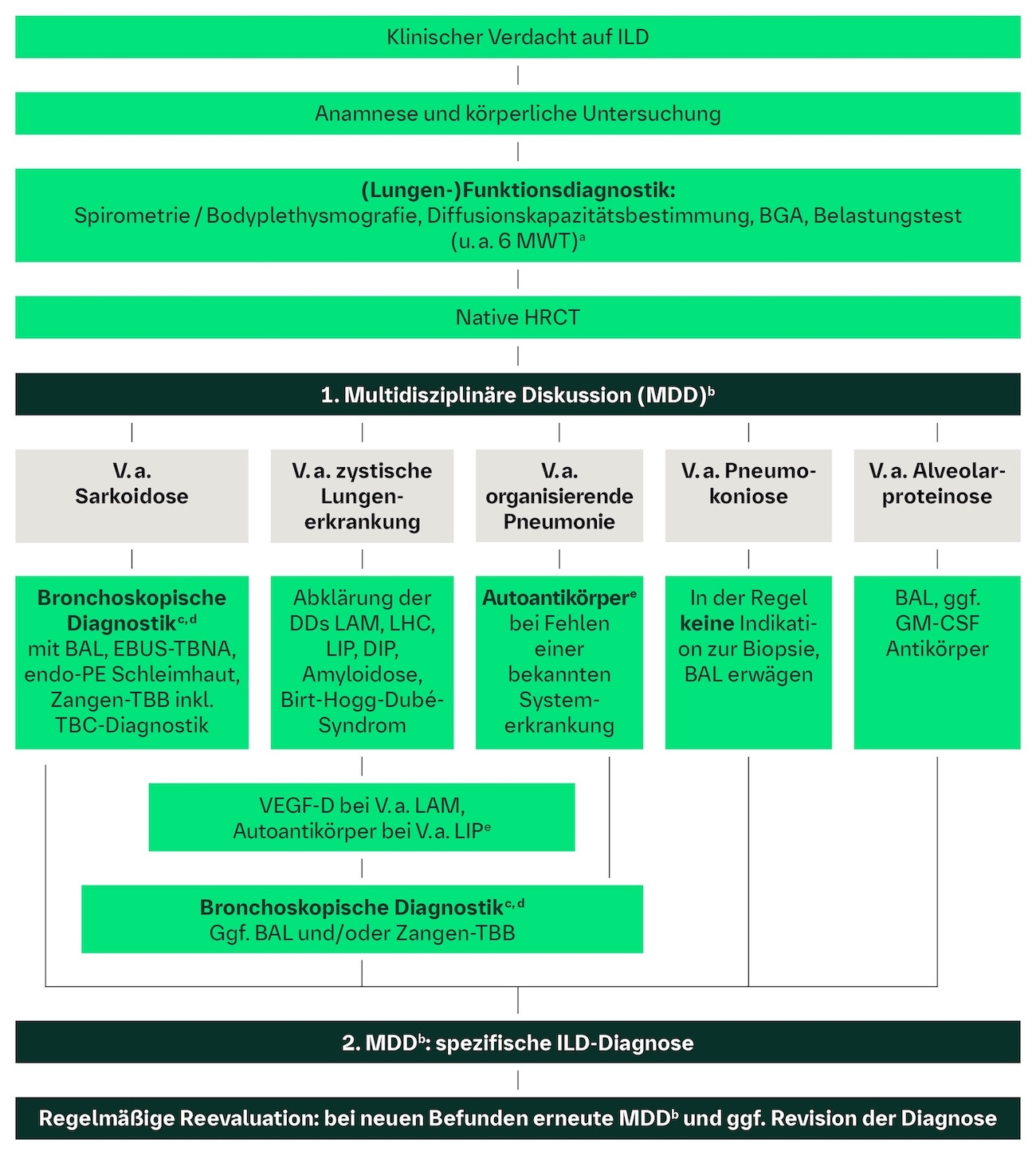
1. Multidisziplinäre Diskussion: Alle vorliegenden Befunde der körperlichen, lungenfunktionellen und radiomorphologischen Untersuchungen sollen multidisziplinär diskutiert (MDD) werden (Fachkräfte aus der Pneumologie, Radiologie, ggf. Pathologie), um über die weiteren diagnostischen Schritte zu entscheiden. Bei ILDs, die mit systemischen Autoimmunerkrankungen assoziiert sind, soll die rheumatologische Fachexpertise mit einbezogen werden.1
4. Autoimmundiagnostik und Serologie1
Nach multidisziplinärer Diskussion wird in Abhängigkeit der ILD-Verdachtsdiagnose eine weiterführende serologische und ggf. bronchoskopische Diagnostik mit transbronchialer Kryobiopsie (Kryo-TBB) und/oder bronchoalveolärer Lavage (BAL) empfohlen.
Die weitere Diagnostik von spezifischen ILDs erfolgt gemäß den jeweiligen aktuellen Leitlinien (z. B. IPF, Lymphangioleiomyomatose, EAA, Sarkoidose).
2. Multidisziplinäre Diskussion: Festlegung der spezifischen ILD-Diagnose. Bei Vorliegen neuer Befunde erneute MDD und ggf. Revision der Diagnose.1
Diagnostik-Algorithmus bei interstitiellen Lungenerkrankungen (ILDs)1
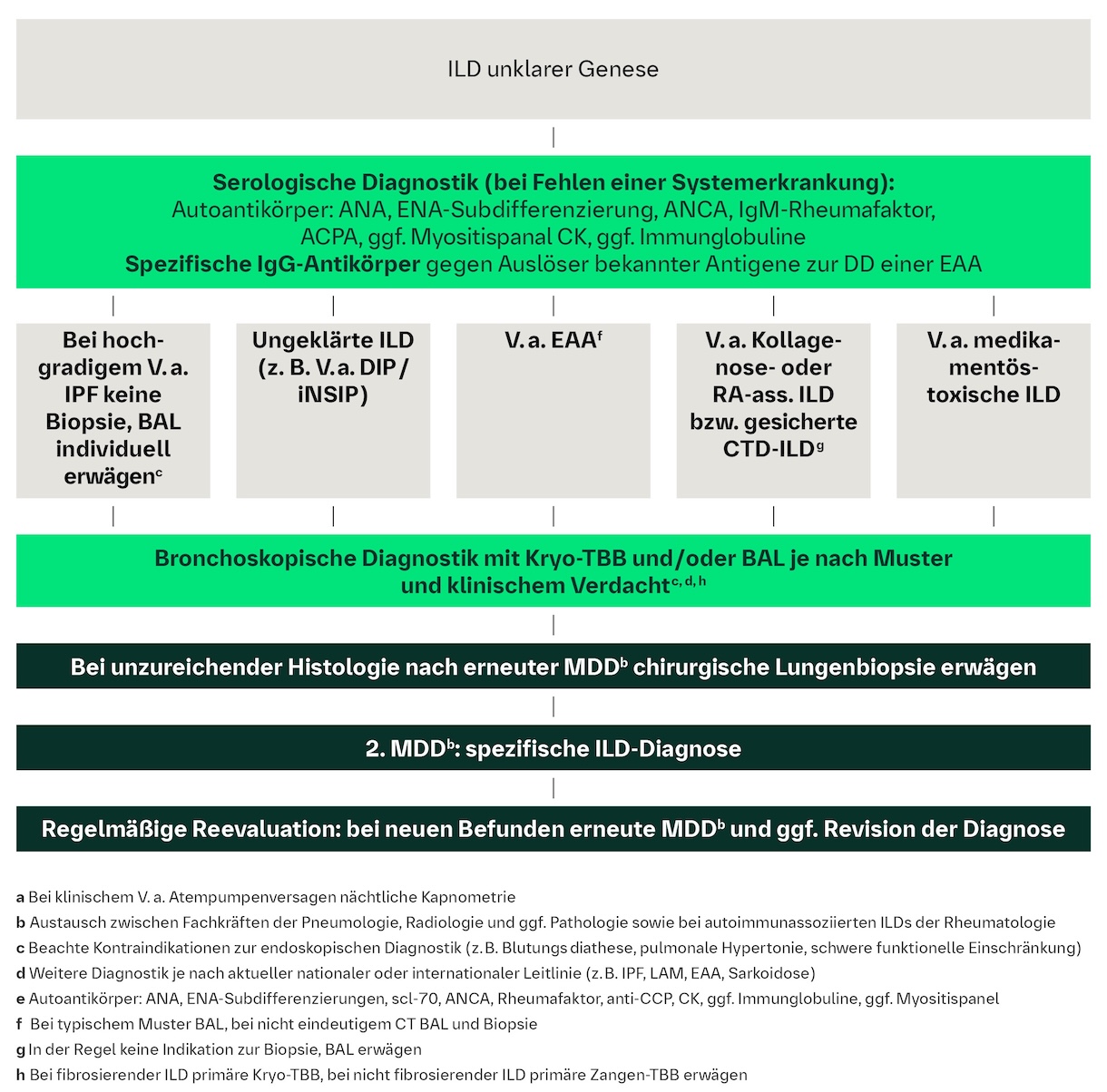
Diagnostik-Algorithmus bei ILD unklarer Genese nach 1. MDD1
Diagnostisches Vorgehen bei Verdacht auf eine progrediente Lungenfibrose (PPF)2
Patient:innen mit ILD weisen ein erhöhtes Risiko für die Entwicklung einer progredienten Lungenfibrose auf. Mit der nationalen S2K-Leitlinie zur Therapie der IPF und anderer progredienter Lungenfibrosen wurden praxisnahe Progredienzkriterien definiert: mindestens 10 % fibrotische Veränderungen im HRCT-Befund, kombiniert mit einer Verschlechterung klinischer, funktioneller oder bildgebender Kriterien innerhalb eines angemessenen Zeitraums (max. 24 Monate).
Nachweis einer progredienten Lungenfibrose3

Insbesondere bei ILD-Patient:innen mit einem erhöhten Progressionsrisiko (höheres Alter, männliches Geschlecht, eingeschränkte Lungenfunktion bei Erstdiagnose, radiologisches UIP-Muster) empfiehlt die S1-Diagnostikleitlinie eine engmaschige klinische und lungenfunktionelle Verlaufskontrolle alle 3–6 Monate. Eine HRCT-Aufnahme kann je nach Verlauf und Risiko alle 12–24 Monate erwogen werden.
Stellenwert der multidisziplinären Disskussion bei Lungenfibrose
Die klinischen, radiologischen und histopathologischen Befunde einer ILD werden in einem multidisziplinären Team / ILD-Board diskutiert. Dieses entscheidet über die Differenzialdiagnose und empfiehlt einen Therapieplan. Vertreten sind Expert:innen aus verschiedenen Disziplinen wie der Pneumologie, Radiologie, Pathologie und bei Verdacht auf autoimmunbedingte ILDs auch Rheumatologie und ggf. Dermatologie. Die Bewertung in einem multidisziplinären Team/ILD-Board ist der Goldstandard bei der ILD-Diagnosefindung, der Beurteilung ihrer Progredienz sowie der Therapieplanung und ist in den deutschen sowie internationalen ILD-Leitlinien verankert.1,2,3,4
Unser Service für Sie: Foliensatz zur S1-Diagnostik-Leitlinie
Der vorliegende Foliensatz fasst die nationale S1-Leitlinie zur Diagnostik von Interstitiellen Lungenerkrankungen (ILDs) im Erwachsenenalter1 zusammen, die interdisziplinär erarbeitet wurde. Er enthält übersichtliche Folien zu den wesentlichen Schritten der ILD-Diagnostik wie: Anamnese, körperliche Untersuchung, Lungenfunktionsdiagnostik, HRCT und spezifische Serologie, invasive Diagnostik mittels BAL und Biopsie sowie zur multidisziplinären Diskussion (MDD).

ERS/EULAR: Europäische Leitlinie zu CTD-ILDs und RA-ILD3
Mit der 2025 veröffentlichten CTD-ILD-Leitlinie legen die European Respiratory Society (ERS) und die European Alliance of Associations for Rheumatology (EULAR) erstmals einen gemeinsamen europäischen Standard für die Diagnostik, das Monitoring und die Therapie interstitieller Lungenerkrankungen im Kontext systemischer Autoimmunerkrankungen vor.3
Ergänzend zur deutschen S1-Diagnostikleitlinie1 empfiehlt die Leitlinie ein systematisches, risikobasiertes Screening auf eine ILD bei allen Betroffenen mit Bindegewebserkrankungen oder einer rheumatoiden Arthritis.3
Screening-Empfehlungen:
- Universell: Bei Systemischer Sklerose (SSc) und Mischkollagenose soll jede Patientin und jeder Patient ab Diagnose ein ILD-Screening mittels HRCT erhalten – unabhängig davon, ob klinische Symptome oder bekannte Risikofaktoren vorliegen.3
- Risikobasiert: Bei Myositis, rheumatoider Arthritis (RA) und Sjögren-Syndrom wird das Screening abhängig von individuellen Risikomerkmalen empfohlen. Diese Risikofaktoren umfassen demografische Daten, klinische Symptome, serologische Befunde und bildgebende Hinweise.3
ILD-Screening-Algorithmus bei systemischen Autoimmunerkrankungen3

Monitoring-Empfehlungen:
Nach einer ILD-Diagnose ist ein strukturiertes und regelmäßiges Monitoring entscheidend, um eine Progression frühzeitig zu erkennen und rechtzeitig therapeutisch gegenzusteuern. So lässt sich das Risiko für Funktionsverlust und Mortalität deutlich senken.
Die CTD-ILD-Leitlinie empfiehlt herfür in den ersten Jahren nach ILD-Diagnose engmaschige Verlaufskontrollen, deren Häufigkeit und Intensität sich nach individuellen Risikofaktoren für Progression und Mortalität richten.3
Risikobasiertes Monitoring bei vorliegender SSc-ILD3

* der Erkrankungsdauer
Risikobasiertes Monitoring bei vorliegender RA-ILD3

* der Erkrankungsdauer

Referenzen
- 1 Kreuter M et al., Pneumologie 2023;77(05):269–302.
- 2 Behr J et al., Pneumologie 2023;77(02):94-119.
- 3 Antoniou KM et al., Eur Respir J. 2025; doi: 10.1183/13993003.02533-2024
- 4 Raghu G et al., Am J Respir Crit Care Med. 2022;205(9):e18–e47.


