
Lungenfibrose-Verlauf: Dramatische Mortalität – Ein Weckruf für die Patientenversorgung.
Die Diagnose Lungenfibrose ist für Patient:innen und Behandelnde oft ein Schock: Als gemeinsames Merkmal vieler interstitieller Lungenerkrankungen (ILD) nimmt sie häufig einen progredient fibrosierenden Verlauf, der die Lungenfunktion zunehmend einschränkt.1 Bei idiopathischer Lungenfibrose (IPF) und progredienter pulmonaler Fibrose (PPF) geht dies mit einer alarmierend hohen Mortalität einher.1,2 Diese Realität verpflichtet zum entschlossenen Handeln. Frühzeitige, differenzierte Diagnostik und eine leitliniengerechte Versorgung nach aktueller Evidenz sind unerlässlich. Denn hinter jeder Statistik stehen Menschen, deren Leben und Lebensqualität auf dem Spiel stehen.
Die hohe Mortalität bei progredienter Lungenfibrose ist eine erschütternde Tatsache.1,2 Unbehandelt schreitet der fibrotische Umbau des Lungengewebes voran, die Sauerstoffaufnahme nimmt ab und das Risiko für einen frühen Tod steigt.1 Diese fortschreitende Verschlechterung beeinflusst die körperliche Leistungsfähigkeit ebenso wie die psychische Stabilität der Betroffenen und ihrer Familien. Für Betroffene, wie Manfred heißt das: Es ist wichtig, die Erkrankung früh zu erkennen, konsequent zu therapieren und interdisziplinär zu begleiten.3
2-Minuten-Check: Ihr Lungenfibrose-Wissen auf dem Prüfstand
Wie hoch ist typischerweise das mediane Überleben bei IPF?
Welcher FVC-Abfall erfüllt ein PPF-Kriterium innerhalb von 24 Monaten?
Wie hoch ist die Mortalität bei einer akuten ILD-Exazerbation?
Was sind die häufigsten Fehldiagnosen bei Lungenfibrose zu Beginn?
Wann ist ein ILD-Board indiziert?
Fakten, die Handeln erfordern – Krankheitsverlauf und Prognose bei Lungenfibrose
Warum ist bei Lungenfibrose keine Zeit zu verlieren? Die Prognose und der typische Krankheitsverlauf liefern klare Antworten.
Das mediane Überleben: Eine ernste Realität
- Idiopathische Lungenfibrose (IPF): Die mediane Überlebenszeit nach der Diagnose beträgt 3 bis 5 Jahre. Damit ist die Prognose ernster als bei vielen Krebserkrankungen.2,4
- Progrediente Lungenfibrose (PPF): Hier variiert das Sterblichkeitsrisiko je nach zugrundeliegender Erkrankung. Die mediane Überlebenszeit bei Patient:innen mit rheumatoider Arthritis und einer Lungenbeteiligung (RA-ILD) liegt bei 3 bis 7 Jahren, bei systemischer Sklerose-assoziierter ILD (SSc-ILD) bei 4 bis 6 Jahren.5,6,7,8 RA-ILD-Patient:innen haben ein 3-fach höheres Sterberisiko.9,10 Mehr als ein Drittel der SSc-bedingten Todesfälle sind auf ILD zurückzuführen, was dies zur häufigsten Todesursache für diese Patient:innen macht.11,12,13,14,15,16
Diese Faktoren können die Mortalität beeinflussen
Alle aufklappen
Das Ausmaß der Fibrose, gemessen durch HRCT, ist ein starker, unabhängiger Prädiktor für die Mortalität sowohl bei IPF als auch bei PPF.
HRCT-Bild mit freundlicher Genehmigung von Priv.-Doz. Dr. Hilmar Kühl

Die Lungenfunktion, gemessen als forcierte Vitalkapazität (FVC), ist einer der wichtigsten Indikatoren für den Krankheitsverlauf. Ein fortschreitender Verlust der FVC steht in direktem Zusammenhang mit einer erhöhten Sterblichkeit.17

Eine akute Exazerbation ist eine plötzliche, dramatische Verschlechterung der Symptome, die oft eine Krankenhauseinweisung erfordert und die Prognose erheblich verschlechtert. Nach einer solchen Krise überlebt nur etwa die Hälfte der IPF-Patient:innen.18 Bei PPF liegt die Mortalität nach einer akuten Exazerbation bei 33 bis 83 %.19 Im Vergleich dazu liegt die Krankenhausmortalität bei einer akuten Exazerbation im Kontext einer COPD bei 11–24 %.20 Das Management dieses Risikos ist daher ein zentraler Bestandteil der Therapie.
Diese Fakten zeichnen ein unmissverständliches Bild. Eine progrediente Lungenfibrose ist schwerwiegend und muss so schnell wie möglich erkannt werden. Die Herausforderung: unspezifische Frühsymptome wie Atemnot oder trockener Husten werden häufig als Anzeichen verbreiteterer Erkrankungen wie COPD oder Herzinsuffizienz fehlgedeutet.21 Außerdem können Begleiterkrankungen die genaue und zeitnahe Diagnose erschweren.22, 23
Jeder Monat, der dadurch verloren geht, ist ein Monat, in dem die Fibrosierung fortschreiten kann und wertvolle Lungenfunktion unwiederbringlich zerstört. Für die Betroffenen bedeutet dies nicht nur eine körperliche, sondern eine immense psychosoziale Last: die Ungewissheit und die Konfrontation mit einer lebenslimitierenden Prognose.1
Frühe Lungenfibrose-Diagnose als Schlüssel – wer, wann, wie im Team?

ILDs sind komplex und ihre frühzeitige, korrekte Diagnose ist entscheidend für die Prognose.24 Aufgrund der heterogenen Krankheitsbilder ist ein isolierter Fachbereich oft nicht ausreichend. Die aktuelle S1-Leitlinie unterstreicht daher die zentrale Bedeutung der interdisziplinären Zusammenarbeit.25
Eine umfassende Anamnese, idealerweise bereits in der hausärztlichen Praxis erhoben, ist ein entscheidender erster Schritt. Gemäß der Diagnostikleitlinie gilt es bei ILDs, mittels Erhebung der Berufsanamnese durch Arbeitseinflüsse verursachte Krankheitsbilder zu erkennen. Dies dient nicht nur dazu, potenziell vermeidbare und ärztlicherseits anzeigepflichtige Berufskrankheiten aufzudecken, sondern auch, um weitere schädliche Expositionen zu eliminieren. Mögliche Expositionen gegenüber Lungenfibrose-auslösenden Substanzen wie Asbest (Asbest-induzierte Lungenfibrose) oder Quarzstaub (Silikose) erfordern eine spezifische Abklärung. Diese Informationen bilden eine wichtige Grundlage für die weitere spezialisierte Diagnostik und die Diskussion im interdisziplinären Team.25
Das Prinzip: ILD-Board als Goldstandard
Die multidisziplinäre Diskussion (MDD), idealerweise im ILD-Board, ist der leitliniengerechte Goldstandard in der Diagnostik.25 Hier werden alle klinischen, radiologischen und ggf. histopathologischen Befunde systematisch zusammengeführt, um eine gesicherte Diagnose zu stellen und die weitere Strategie festzulegen.
Die wichtigsten Fachbereiche im ILD-Board25
Pneumologie: Führt die klinische Bewertung durch, bringt die Anamnese ein und steuert den diagnostischen Prozess.
Radiologie: Ist verantwortlich für die Interpretation der hochauflösenden Computertomografie (HRCT), die als Goldstandard für die ILD-Diagnose gilt.
Rheumatologie: Spielt eine entscheidende Rolle bei der Abklärung, ob eine rheumatische Systemerkrankung die Ursache der ILD ist.

![Wir Radiologen müssen den Austausch mit den überweisenden Ärzten pflegen. Wir müssen uns einmischen. Wir sind nicht nur eine Art Toröffner durch Zufallsbefunde, sondern im Grunde genommen [...] können wir durchaus eine Art Game-Changer sein. Dr. med. Beate Rehbock (Fachärztin für Diagnostische Radiologie, Berlin)](/de/sites/default/files/2026-01/Carousel_Zitat_Dr_Rehbock_Radiologin.jpg)

Je nach Befundlage wird das Kernteam durch weitere Expertisen ergänzt (z. B. Pathologie). Im Zusammenspiel dieser Disziplinen entstehen präzise Diagnosen und tragfähige Therapieentscheidungen.25
Unser Tipp für Sie: De Zeeuw, Schäfer und die Hürden und Chancen bei ILD-Boards

Unterhaltung und gleichzeitig informieren? Dann sind Sie bei unserem Pneumo-Podcast genau richtig.
Jetzt reinhören
Leitlinien-Navigator – Handlungsempfehlungen für die Praxis
Leitlinien sind das Fundament der evidenzbasierten Medizin, doch ihre Anwendung im häufig dicht getakteten Praxisalltag kann herausfordernd sein. Der Leitlinien-Navigator wurde entwickelt, um die wichtigsten Empfehlungen zu ILDs für Sie aufzubereiten.
Er übersetzt die Empfehlungen der ERS/EULAR-Leitlinie zu CTD-ILDs, der S2K-Diagnostikleitlinie bei IPF, der S1-Diagnostikleitlinie bei ILDs sowie der S2K-Therapieleitlinie bei IPF und PPF in vier interaktive, praxisorientierte Module. Ziel ist es, Ihnen schnell und strukturiert zu helfen, das individuelle Risiko Ihrer Patient:innen besser einzuschätzen, eine Progression frühzeitig zu erkennen und die nächsten Schritte gezielt zu planen. Jedes Modul führt Sie in wenigen Minuten durch die entscheidenden Kriterien und liefert Ihnen eine zusammenfassende Handlungsempfehlung.

Monitoring
von ILDs
Empfehlungen der ERS/EULAR- Leitlinie und der S1-ILD-Diagnostikleitlinie
Jetzt startenPPF-Kriterien-Check
(Nachweis einer
Progredienz)
Empfehlungen der S2K-Therapieleitlinie bei IPF und PPF
Jetzt startenILD-Screening bei CTD und RA
Empfehlungen der ERS/EULAR-Leitlinie zu CTD-ILDs
Da Patient:innen mit Bindegewebserkrankungen (CTD) eine ILD entwickeln können, ist ein frühzeitiges Screening entscheidend. Dieses Modul fasst die Empfehlungen der ERS/EULAR-Leitlinie zu CTD-ILDs zusammen.
Alle Patient:innen mit SSc sollten mittels HRCT auf ILD gescreent werden (starke Empfehlung)
Alle Patient:innen mit MCTD sollten mittels HRCT auf ILD gescreent werden (starke Empfehlung)
- Alle Patient:innen mit IIM sollten mittels HRCT auf ILD gescreent werden (bedingte Empfehlung)
- Starke Empfehlung: Bei Vorliegen von Risikofaktoren Patient:innen mit IIM mittels HRCT auf ILD screenen
Liegt einer oder mehrere dieser Risikofaktoren vor?
- Höheres Alter
- Männliches Geschlecht
- Rauchen
- Erhöhte ESR
- Vorhandensein von anti-CCP, RF
- Höhere Gelenkerkrankungsaktivität
Liegt einer oder mehrere dieser Risikofaktoren vor?
- Höheres Alter
- Männliches Geschlecht
- Erhöhtes CRP
- Vorhandensein von anti-Ro52
- Extrapulmonale Beteiligung
IPF-Diagnose
Empfehlungen der nationalen S2K-Leitlinie zu IPF
Die idiopathische Lungenfibrose (IPF) ist eine fortschreitende Erkrankung ist, bei der eine schnelle und adäquate Diagnosestellung entscheidend ist. Dieses Modul fasst die Empfehlungen der nationalen S2KLeitlinie zur IPF-Diagnostik zusammen.
Zum Diagnose-AlgorithmusDifferenzierter IPF-Diagnosealgorithmus nach S2K-Leitlinie
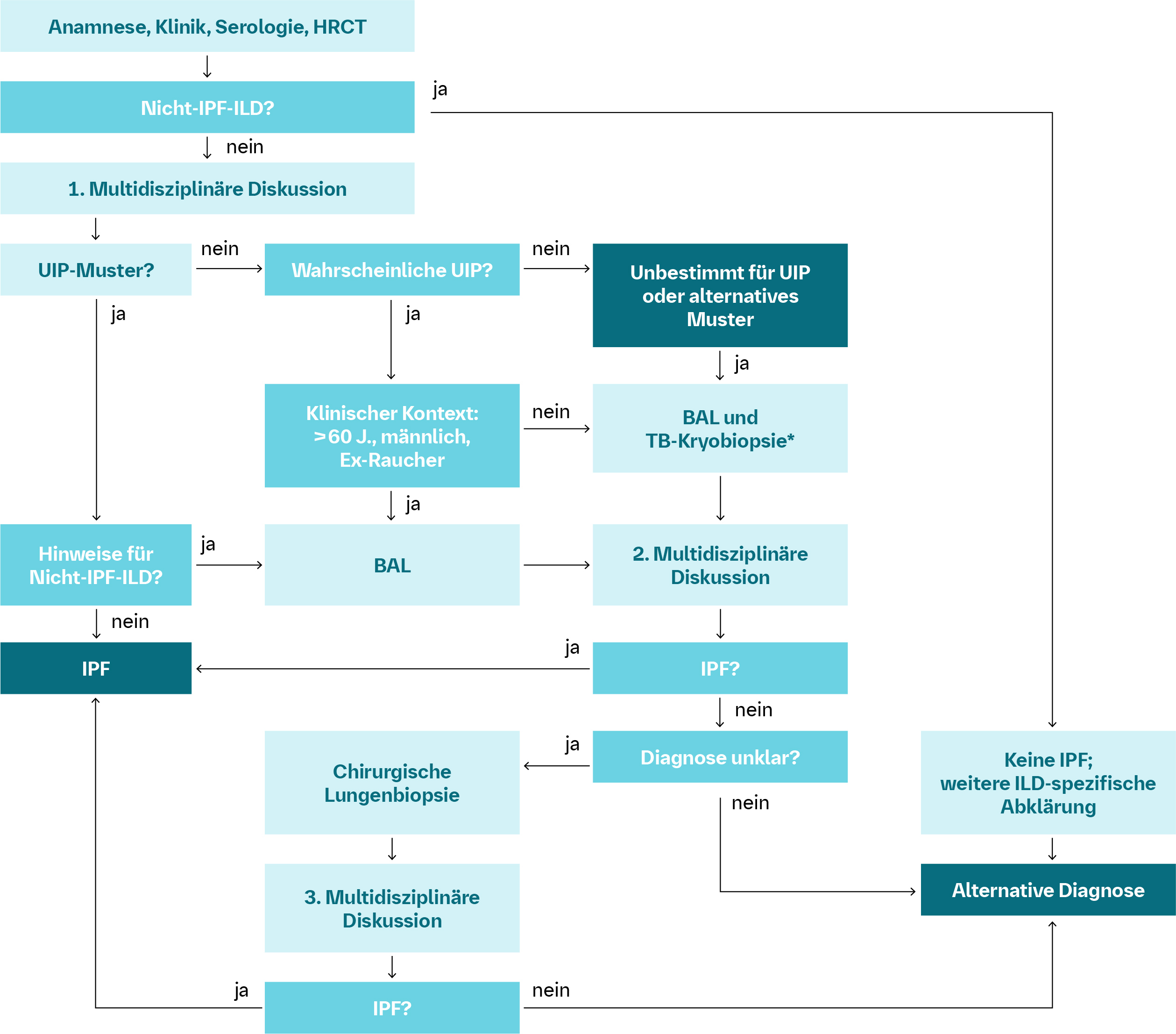
Für die IPF-Diagnose müssen folgende zwei Kriterien erfüllt sein:
- Ausschluss einer ILD bekannter Ursache und der Nachweis eines UIP-Musters im HRCT oder
- Ausschluss einer ILD bekannter Ursache und eine spezifische Kombination von HRCT und Histologie
Monitoring von ILDs
Empfehlungen der ERS/EULAR-Leitlinie und der S1-ILD-Diagnostikleitlinie
Ein erheblicher Teil von ILDs verläuft progredient. Daher ist ein engmaschiges Monitoring bei ILDs unerlässlich, um den Krankheitsverlauf zu verfolgen und eine Verschlechterung frühzeitig zu erkennen.
Monitoring bei autoimmunen ILDs
Empfehlungen der ERS/EULAR-Leitlinie
Diese Faktoren gehen mit einem erhöhten Progressionsrisiko einher:
• Höheres Alter, männliches Geschlecht
• Afroamerikanische Ethnie
• Erhöhte ESR/CRP
• Positiv auf ATA-I
• Größeres Ausmaß der ILD im HRCT
• Hautbeteiligung (höheres Ausmaß und Progression)
Diese Faktoren gehen mit einem erhöhten Progressionsrisiko einher:
• Höheres Alter bei Krankheitsbeginn
• Männliches Geschlecht
• Anti-CCP Antikörper
• Rheumafaktor
• Niedrige FVC und/oder DLCO bei Baseline
• UIP/wahrscheinliches UIP-Muster
• Größeres Ausmaß der ILD im HRCT
• Stärkere Gelenkbeteiligung

Diese Faktoren gehen mit einem erhöhten Progressionsrisiko einher:
• Erhöhtes Ferritin
• Anti-MDA-5, Anti-Synthetase
• Größeres Ausmaß der ILD und ILD-Muster im HRCT

(z. B. bei Sjögren-Syndrom oder Mischkollagenose) ▼

Antoniou KM et al., Eur Respir J. 2025; in press: 2500896.
ILD-Monitoring (allgemein)
Empfehlungen der S1-ILD-Diagnostikleitlinie
-> Klinisches und lungenfunktionelles Monitoring bei ILD alle 3–6 Monate*
6 MWT: alle 12 Monate erwägen, HRCT: je nach Verlauf und Risiko, ggf. alle 12–24 Monate erwägen
* bei Patienten mit niedrigem Progressionsrisiko (z. B. milde Erkrankung, langer bestehende ILD) ggf. längerfristige Kontrollen. Erhöhtes Progressionsrisiko: Alter, männliches Geschlecht, eingeschränkte Lungenfunktion (FVC; DLCO) bei Erstdiagnose, radiologisches UIP Muster.
Monitoring-Algorithmus
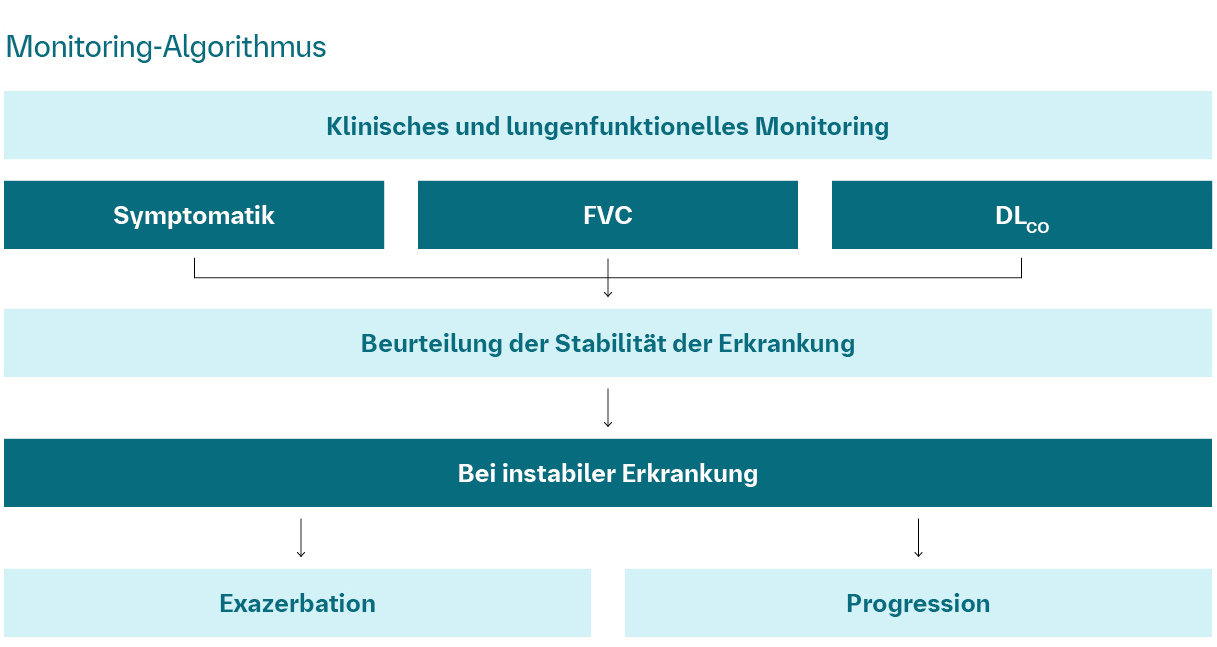
Kreuter M et al., Pneumologie 2023;77(05):269–302.
PPF-Kriterien-Check (Nachweis einer Progredienz)
Empfehlungen der S2K-Therapieleitlinie bei IPF und PPF
Die Entscheidung, ob eine ILD eine progrediente Lungenfibrose (PPF) aufweist, ist wegweisend für die weitere Therapie. Die S2K-Leitlinie definiert hierfür klare, praxisnahe Kriterien.
 HRCT-Befund mit mind. 10 % fibrotischen Veränderungen liegt vor
HRCT-Befund mit mind. 10 % fibrotischen Veränderungen liegt vor
und
Ein relativer Abfall der FVC ≥ 10 % v. Soll
oder Nachweis von mindestens zwei der folgenden Kriterien:
-
Zunahme der respiratorischen Symptome
-
Zunahme der Fibrosezeichen im HRCT *
-
Langzeitsauerstofftherapie
-
Relativer FVC-Abfall von ≥ 5 % v. Soll
-
Absoluter Abfall der DLCO um ≥ 15 % v. Soll
-
Akute ILD-Exazerbation mit Hospitalisation
-
Abnahme der Gehstrecke im 6-Minuten-Gehtest um ≥ 50 m oder 20 % und/oder Abnahme der
minimalen Sauerstoffsättigung während des 6-Minuten-Gehtests um > 5 % und unter 88 % absolut
Ergebnis
 PPF-Kriterien noch nicht erfüllt → Weiterhin engmaschiges Monitoring empfohlen.
PPF-Kriterien noch nicht erfüllt → Weiterhin engmaschiges Monitoring empfohlen.
Ergebnis
 PPF-Kriterien erfüllt → Empfehlung: interdisziplinär validieren (idealerweise im ILD-Board);
antifibrotische Therapie erwägen
PPF-Kriterien erfüllt → Empfehlung: interdisziplinär validieren (idealerweise im ILD-Board);
antifibrotische Therapie erwägen
* Radiologische Fibrosezeichen definiert als: Zunahme von Ausmaß oder Schweregrad von Traktionsbronchi(ol)ektasen | Neu auftretende Milchglasverschattung mit Traktionsbronchiektasen
| Neu auftretende feine Retikulationen | Zunahme von Ausmaß oder Deutlichkeit von Retikulationen | Neuauftreten oder Zunahme von Honigwaben |
Zunehmender Volumenverlust von Lungenlappen
Behr J et al., Pneumologie 2023;77(02):94–119.
ILD-Leitlinien kompakt zusammengefasst
Nach Login auf unserer Fachkreis-Seite erhalten Sie die wichtigsten Informationen der ILD-Leitlinien kompakt zusammengefasst und bereit zum Download.
Melden Sie sich jetzt für Ihre passende Fachgruppe an:
Effektivität der aktuellen Lungenfibrose-Therapie
Antifibrotische Therapien sind derzeit die einzigen zugelassenen medikamentösen Optionen, die das Fortschreiten der Fibrose verlangsamen können. Sie reduzieren den jährlichen FVC-Verlust signifikant und senken das Risiko akuter Exazerbationen.3
Weitere Therapieoptionen bei Lungenfibrose:26,27,28,29
- Sauerstofftherapie bei Hypoxämie zur Verbesserung der Belastbarkeit
- Pulmonale Rehabilitation zur Stabilisierung der körperlichen Leistungsfähigkeit
- Lungentransplantation als Option bei fortgeschrittener Erkrankung und geeigneter Patientenselektion
- Optimiertes Management der Grunderkrankung bei autoimmunassoziierten ILDs
Trotz dieser Möglichkeiten zeigen sich in der Praxis drei zentrale Herausforderungen:
- Die späte Diagnosestellung: Wird die Diagnose erst spät gestellt, geht Lungenfunktion verloren, die nicht wieder hergestellt werden kann.24
- Therapiemanagement und Nebenwirkungen: Unerwünschte Wirkungen können die Adhärenz gefährden und zu Therapieabbrüchen führen. Ein proaktives Management ist hier unerlässlich.30
- Die uneinheitliche Umsetzung der Leitlinien: Fehlendes Monitoring und der verzögerte Einsatz einer adäquaten antifibrotischen Therapie widersprechen der Evidenz und gefährden die Versorgung von Patient:innen mit Lungenfibrose.
Wie die Versorgung bei Lungenfibrose verbessern? 5 Fragen – 5 Antworten





FAQ: Häufig gestellte Fragen zum Verlauf der Lungenfibrose
Im Praxisalltag tauchen immer wieder ähnliche Fragen zur Mortalität der Lungenfibrose auf. Hier finden Sie kompakte Antworten, die Sie direkt in der Betreuung Ihrer Patient:innen unterstützen können.
Weil es sich um progrediente, irreversible Erkrankungen handelt, die unbehandelt zu respiratorischem Versagen führen können.1
Direkte Mortalitätsdaten sind limitiert, aber Studien zeigen eine signifikante Reduktion von Progression und Exazerbationen – beides Schlüsselfaktoren für das Überleben.3
Sie ist die einzige potenziell kurative Option, aber nur für ausgewählte Patient:innen geeignet.28
Durch rechtzeitige Therapieeinleitung, Infektionsprophylaxe, Patientenschulung und engmaschiges Monitoring.31,32,33
Wissen vertiefen, Versorgung bei Lungenfibrose optimieren
Die richtigen Informationen sind der Schlüssel, um Patient:innen mit Lungenfibrose optimal zu versorgen. Hier finden Sie alles, was Sie für einen umfassenden Überblick und ein erfolgreiches Therapiemanagement bei Lungenfibrose benötigen.


Fachinformationen zu Lungenfibrose




Leben mit Lungenfibrose – Informationen für Patient:innen und Angehörige
Ein aktives und erfülltes Leben mit Lungenfibrose ist trotz der Erkrankung möglich. Wichtig für Betroffene ist es, den Alltag bewusst zu gestalten und Strategien zu entwickeln, die helfen, mit den Symptomen umzugehen und das Wohlbefinden zu fördern.
Glossar: Fachbegriffe rund um IPF und PPF
Alle aufklappenPlötzliche, unerklärte Verschlechterung der respiratorischen Symptome und Lungenfunktion bei ILD.
Forcierte Vitalkapazität – zentraler Parameter zur Beurteilung der Lungenfunktion.
Interstitielle Lungenerkrankung im Rahmen systemischer Autoimmunerkrankungen.
Anmelden oder Registrieren
Dieser Inhalt ist exklusiv für Fachkreise und Behandlungsteams. Jetzt kostenfrei anmelden oder registrieren und uneingeschränkten Zugriff auf unsere Services erhalten:
- Alle Informationen zu Produkten und Therapiegebieten
- Aktuelle Kongressberichte
- Neue Studiendaten und Literatur
- Materialien zur Patientenaufklärung
- Zahlreiche Downloads und Bestellmaterialien
- Bilddatenbank und Foliensätze
Referenzen
- 1 Cottin V et al. Eur Respir Rev 2018;27(150):pii:180076.
- 2 Vancheri C et al. Eur Respir J. 2010;35:496–504.
- 3 Behr J et al., Pneumologie 2023;77(02):94–119.
- 4 Martinez FJ et al., Nat Rev Dis Primers. 2017;3:17074.
- 5 Juge PA et al. RMD Open. 2023;9(4):e003491.
- 6 Hyldgaard C et al. Respirology. 2017;22(3):494–500.
- 7 Moore OA et al. Rheumatology (Oxford). 2013;52(1):155–60.
- 8 Hu S et al. Arthritis Res Ther. 2018;20:235.
- 9 Olson AL et al., Am J Respir Crit Care Med. 2011;183(3):372–78.
- 10 Hyldgaard C et al., Respiration. 2019;98(5):455–60.
- 11 Cappelli S et al. Eur Respir Rev. 2015;24(137):411–19.
- 12 Distler O et al. Eur Respir J 2020;55(5):1902026.
- 13 Distler O et al. Expert Rev Clin Immunol. 2019;15(10):1009–17.
- 14 Tyndall AJ et al. Ann Rheum Dis. 2010;69(10):1809–15.
- 15 Steen VD and Medsger TA. Ann Rheum Dis. 2007;66(7):940–44.
- 16 Roofeh D et al. Curr Opin Rheumatol. 2019;31(3):241–49.
- 17 Maher TM et al., Respirology. 2023;10.1111/resp.14579.
- 18 Song JW et al., Eur Respir J. 2011;37(2):356–363.
- 19 Kolb M et al., Eur Respir Rev. 2018;27(150):180071.
- 20 Anzueto A 2010. Eur Respir Rev 2010 19(116):113–18.
- 21 Cosgrove GP et al., BMC Pulm Med. 2018; 18:9.
- 22 Koudstaal T et al. Trends Mol Med. 2023;29(12):1076–87.
- 23 Luppi F et al. Respir Res. 2021;22(1):109.
- 24 Cano-Jiménez E et al. Sci Rep. 2021;11(1):9184.
- 25 Kreuter M et al., Pneumologie 2023;77(05):269–302.
- 26 Koudstaal T, Wijsenbeek MS. Presse Med. 2023;52(3):104166.
- 27 Swigris JJ et al., Respir Care. 2011;56(6):783–89.
- 28 Strykowski R, Adegunsoye A. Immunol Allergy Clin North Am. 2023;43(2):209-28.
- 29 Wijsenbeek M, Cottin V. N Engl J Med. 2020;383(10):958-68.
- 30 Burnier, Michel. Eur J Intern Med. 2024;119:1-5.
- 31 Kolb M et al. Eur Respir Rev. 2018;27(150):180071.
- 32 Richeldi L et al., Respir Med 2016;113:74–79.
- 33 Flaherty KR et al., N Engl J Med. 2019;381(18):1718–27.